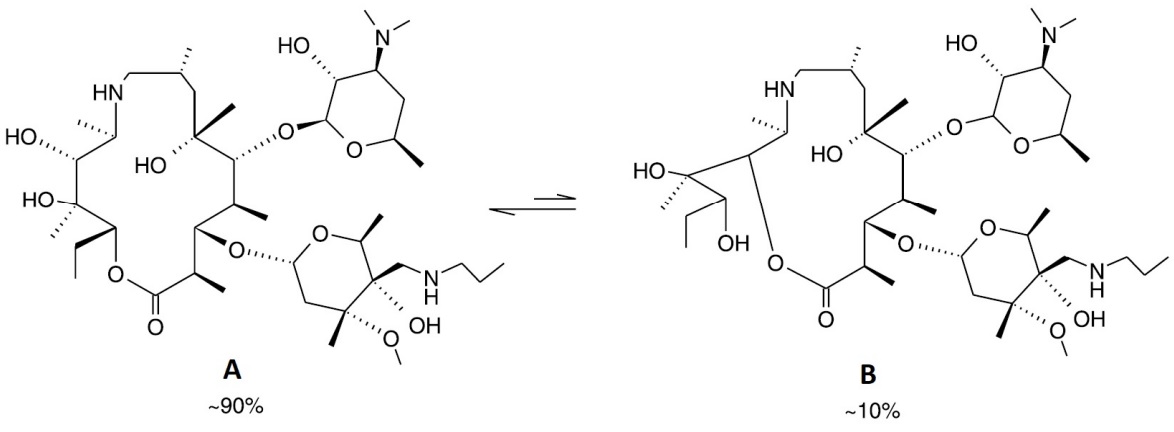
Figura 1. Estructura química de los isómeros de tulatromicina. Fuente: J. vet. Pharmacol. Therap. 2004; 27(4), 203–101 .
ARTÍCULO DE INVESTIGACIÓN
Validación de un método para cuantificar tulatromicina en tejidos bovinos mediante cromatografía líquida de alta performance con detección por espectrometría de masas
Barcarolo, D1; Addona SM1,3; Angeli, E1,2; Peralta, MB1 ; Ortega, HH1,2; Hein, GJ1,4
1
Laboratorio de Biología Celular y Molecular Aplicada, ICiVet-Litoral (UNL-CONICET), Esperanza, Santa Fe, Argentina.
2
Facultad
de Ciencias Veterinarias, Universidad Nacional del Litoral, Esperanza, Santa Fe, Argentina.
3
Facultad de Ingeniería Química,
Universidad Nacional del Litoral, Santa Fe, Argentina.
4
Centro Universitario Gálvez (CUG-UNL), Gálvez, Santa Fe, Argentina.
Recibido: 09/08/2021
Aceptado: 01/02/2022
Correspondencia e-mail:Gustavo Juan Hein ghein@santafe-conicet.gov.ar
Resumen
La tulatromicina es un antimicrobiano aprobado para el tratamiento de enfermedades respiratorias de bovinos. Dada la importancia del uso de este antibiótico en los animales, se validó un método analítico basado en cromatografía liquida de alta performance en tandem con ionización por electrospray y detección por espectrometría de masas/masas (LC ESI MS/MS) para cuantificar tulatromicina en diferentes tejidos bovinos, de acuerdo a normas de Agencias Regulatorias Nacionales e Internacionales. El procedimiento optimizado implicó la homogeneización de la muestra con acetato de etilo y ácido clorhídrico, e hidrólisis de la fase acuosa resultante, para convertir la tulatromicina y sus metabolitos en el residuo marcador (2R, 3S, 4R, 5R, 8R, 10R, 11R, 12S, 13S, 14R) -2-etil-3,4,10,13-tetrahidroxi-3, 5,8,10,12,14-hexametil-11 – [[3,4,6-tridesoxi-3- (dimetilamino) -β-D-xilohexopiranosil] oxi] -1-oxa-6-azaciclopentadecan-15-ona, conocido como CP-60,300. Seguidamente, se realizó una limpieza del extracto acuoso mediante un cartucho de intercambio catiónico y posterior análisis por LC ESI MS/MS. La metodología analítica propuesta fue validada cumpliéndose los criterios de aceptación establecidos por los organismos de referencia en la temática, demostrando ser adecuada para el análisis de residuos de tulatromicina en tejidos bovinos con fines reglamentarios.
Palabras clave: CP-60,300, hidrólisis, validación, tejidos
Validation of analytical methodology to quantify tulathromycin in bovine tissues by high performance liquid chromatography and detection by mass spectrometry
Summary
Tulathromycin is an approved antimicrobial used in cattle to treat respiratory diseases. An analytical method based on high performance liquid chromatography in tandem with electrospray ionization and detection by mass / mass spectrometry (LC ESI MS / MS) was validated to quantify tulathromycin in different bovine tissues according to standards of National and International Regulatory Agencies. The optimized procedure involved homogenizing the sample with ethyl acetate and hydrochloric acid, hydrolysis of the resulting aqueous layer to convert tulathromycin and its metabolites into the marker residue (2R, 3S, 4R, 5R, 8R, 10R, 11R, 12S, 13S, 14R) -2-etil-3,4,10,13-tetrahidroxi-3, 5,8,10,12,14-hexametil-11 – [[3,4,6-tridesoxi-3- (dimetilamino) -β-D-xilohexopiranosil] oxi] -1-oxa-6-azaciclopentadecan-15-ona, also known as CP-60,300, cleaning by using a cation exchange cartridge and subsequent analysis by LC ESI MS/MS. The proposed analytical methodology was validated meeting the acceptance criteria established by the reference organisms, proving to be adequate for the analysis of tulathromycin residues in bovine tissues for regulatory purposes.
Key words: CP-60,300, hydrolysis, validation, tissues
INTRODUCCIÓN
La tulatromicina es un agente antimicrobiano macrólido semisintético, perteneciente a una serie de antibióticos macrocíclicos tribásicos que se han denominado triamilidas. Se
ha desarrollado para la administración parenteral única en bovinos y tiene actividad in vitro
contra: Mannheimia haemolytica, Pasteurella
multocida y Haemophilus somnus y Actinobacillus pleuropneumoniae, Pasteurella multocida y
Mycoplasma hyopneumoniae, que son las bacterias patógenas más comúnmente asociadas a
las enfermedades respiratorias del ganado bovino1,12. La tulatromicina en medio acuoso, consiste en una mezcla equilibrada de dos isómeros: A
y B; en una relación porcentual 90:10, respectivamente (Figura 1)2.
La administración en especies animales
destinadas al consumo humano, requiere de métodos de vigilancia reglamentaria de los residuos
de tulatromicina en los tejidos comestibles de los
animales de producción. En este sentido, los organismos reguladores de cada país, establecen cuales son los límites máximos de residuos (LMR)
permitidos para asegurar la calidad de los alimentos. En Argentina, el Servicio Nacional de Sanidad y Calidad Agroalimentaria (SENASA) es la
entidad responsable de reglamentar los LMR en
alimentos de origen animal, mediante la resolución número (N°) 559 del año 201111. Dichos LMR
resultan necesarios para establecer los períodos
de restricción de los productos veterinarios, que
se deben respetar para que los alimentos provenientes de animales tratados resulten seguros
y no generen inconvenientes a la salud pública,
siendo para bovinos (mg/kg): grasa 0,1, hígado y
riñón 39
. En cuanto a la tulatromicina, se metaboliza en varios compuestos, siendo el principal residuo marcador el metabolito (2R, 3S, 4R, 5R, 8R,
10R, 11R, 12S, 13S, 14R) -2-etil-3,4,10,13-tetrahidroxi-3, 5,8,10,12,14-hexametil-11 – [[3,4,6-tridesoxi-3- (dimetilamino) -β-D-xilohexopiranosil]
oxi] -1-oxa-6-azaciclopentadecan-15-ona, también conocido como CP-60,300, producto de la hidrólisis de la tulatromicina y sus metabolitos12,15.
Es así, que todas las guías establecen como residuo marcador al metabolito CP-60,300 para establecer el LMR5,8,9.
El objetivo de este trabajo fue validar una
metodología analítica para identificar y cuantificar tulatromicina en diferentes tejidos de bovinos: hígado, riñón, músculo, sitio de inyección y
grasa, por LC ESI MS/MS.
Figura 1. Estructura química de los isómeros de tulatromicina. Fuente: J. vet. Pharmacol. Therap. 2004; 27(4), 203–101
.
MATERIALES Y MÉTODOS
En el presente estudio, se utilizaron muestras de tejidos bovinos: hígado, riñón, músculo, sitio de inyección y grasa, provenientes de animales de frigoríficos faenados. Para la determinación analítica del compuesto en estudio se utilizó como estándar interno (EI), tilmicosina fosfato (Til), otro antibiótico de la familia de los macrólidos, para normalizar las pérdidas del compuesto de interés durante el tratamiento de muestras. El estándar de Til es de la firma FIPHARM Co. Limited, China y el de tulatromicina de LIVZON New North River Pharmaceutical co. Ltd., China. Se tomó como referencia el trabajo publicado por Saito-Shida et al. (2019)12, y a partir del mismo se realizaron una serie de modificaciones y se incorporaron nuevas matrices.
Tratamiento de muestras
Se pesó 1 g de tejido representativo de la muestra, previamente disgregado, y se fortificó con 0,1 mL de solución de trabajo de Til nominal 100 mg/L para las matrices hígado, riñón y sitio de inyección, y nominal de 10 mg/L para músculo y grasa. Junto al EI, se adicionó 0,1 mL de soluciones de trabajo de concentraciones diferentes de tulatromicina en el rango analítico propuesto para cada matriz. Se homogenizó durante 1 min con 2,5 mL de acetato de etilo y luego se agregaron 2,5 mL de HCl, concentración nominal 2 M, y se homogenizó 1 min. Se sonicó 10 min y luego se centrifugó a 3000 g x durante 10 min, descartándose la fase orgánica y separando la fase acuosa en otro tubo (primer extracto). Se repitió la operación con el pellet de tejido, agregando 1 mL de acetato de etilo y luego 1 mL de HCl, para luego descartar la fase orgánica y combinar la fase acuosa obtenida con la de la primera extracción. Se llevó a un baño termostatizado a 60 °C durante 0,5 h para la hidrólisis. Posteriormente, se realizó una limpieza mediante extracción en fase sólida (SPE) con un cartucho de intercambio catiónico STRATA XC 33 µm 60 mg/3mL, para lo cual se acondicionó el lecho con 1 mL de metanol y 2,5 mL de agua; se dispensaron 0,5 mL del extracto acuoso, se lavó con 0,5 mL de metanol y se eluyó con 0,5 mL de una solución de hidróxido de amonio/ metanol (5:95 %). El eluato se evaporó bajo corriente de nitrógeno, en un baño termostatizado a 50 °C durante 5 min y posteriormente se reconstituyó en 150 µL de metanol/agua (1:1), para luego analizar mediante inyección directa en el instrumento.
Equipamiento
Se utilizó un cromatógrafo de líquidos UFLC Shimadzu que consta de dos bombas binarias, un desgasificador y un muestreador automático SIL-20AC XR con horno para calentamiento de columna. La separación cromatográfica se realizó en una columna analítica Shimadzu Shim-pack GIST C18 (3,0 µm, 100 mm x 4,0 mm) provista de una columna de protección Shimadzu-pack GIST (G) C18 (3,0 µm, 4,0 x 10 mm) y termostatizada a 35 °C. La identificación y cuantificación se logró mediante el uso de un espectrómetro de masas triple cuadrupolo QTrap 3200 (AB Sciex) equipado con una fuente de iones por electrospray (ESI).
Condiciones operativas del cromatógrafo de líquidos y espectrómetro de masas
La fase móvil A consistió en una solución buffer de acetato de amonio nominal 10 mM, pH 4, con ácido acético glacial, y la fase móvil B consistió en metanol. La velocidad de flujo fue de 0,35 mL/min y el volumen de inyección de 5 μL. El gradiente se inició con 50 % de B durante 4 min, modificándose hasta el 100 % en 0,5 min y luego se mantuvo durante otros 0,5 min. Seguidamente, se regresó al 50 % en 0,5 min y se mantuvo durante otros 4,5 min para lograr reequilibrar la columna. El espectrómetro de masas triple cuadrupolo operó en modo ionización positiva, y la identificación y cuantificación se realizó mediante monitoreo de reacciones múltiples (MRM). El gas cortina fue seteado en 20 psi, el gas de nebulización (GS1) y el gas de secado (GS2) en 60 y 50 psi respectivamente, el voltaje de ionización por electrospray fue de 5500 V y la temperatura de la fuente de 650 °C. Los parámetros optimizados para cada ion se muestran en la tabla 1.
Validación de la metodología analítica para bovinos
La validación se define como el proceso
de asegurar que un método analítico sea capaz
de brindar resultados confiables con precisión y
exactitud aceptables. Para validar la metodología analítica, se estudiaron las cifras de mérito,
tomándose como referencia distintos organismos de referencia en la temática3,7,10,13.
Selectividad/Especificidad. Para diferenciar unívocamente el compuesto de interés en
presencia de otros componentes en la matriz
de muestra bajo las condiciones de análisis
establecidas, se analizaron dos extractos de:
solvente, soluciones de trabajo del compuesto
en estudio y estándar interno (EI), extractos
blancos de matriz procesados y extractos
blancos de matriz procesados con el agregado
del compuesto en estudio y EI. Criterio de
aceptación: ausencia de interferencia entre
la señal del compuesto en estudio/EI y las
señales del disolvente y/o de la matriz; en
caso de interferencias:
Tabla 1. Principales parámetros optimizados para CP-60,300 y Til.
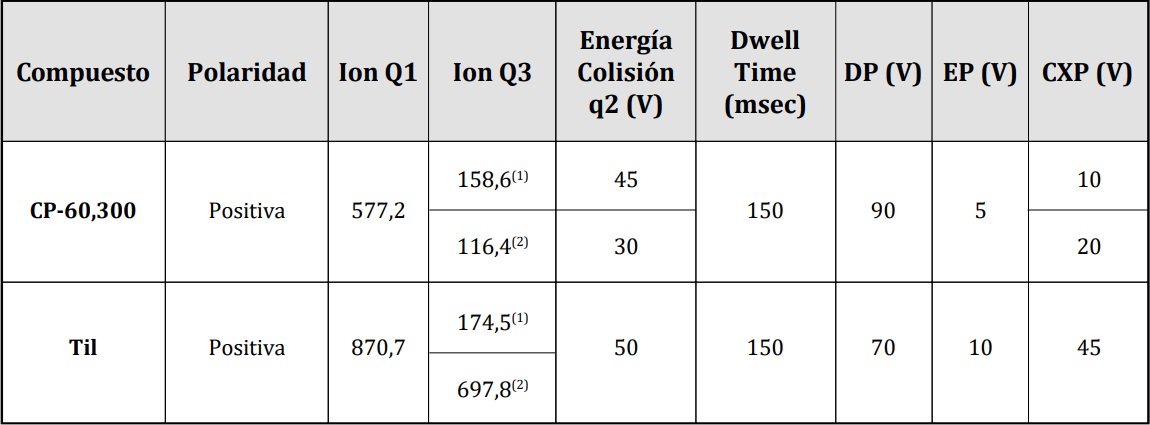
(1): ion de cuantificación; (2): ion de confirmación; Q1, q2 y Q3: cuadrupolos del espectrómetro de masas; Dwell Time: tiempo del experimento (transición), milisegundos (msec); DP: voltaje en el orificio (Declustering Potencial), Voltio (V); EP:
potencial de entrada (Entrance Potential), V; CXP: Potencial de salida de la celda de colisión (Collision Cell Exit Potential), V.
• La señal del compuesto interferente en el
tiempo de retención del compuesto de interés, debe ser inferior al 20 % de la respuesta del compuesto de interés en el límite de
cuantificación.
• La señal del compuesto interferente en el
tiempo de retención del EI, debe ser inferior
al 5 % de la respuesta del EI.
Linealidad y rango. La curva de calibración se preparó en tejido, a partir de fortificar
un pool de tejido testigo con soluciones de trabajo del compuesto en estudio (tulatromicina)
y el EI (Til), y posterior procesamiento según
tratamiento de muestra descripto anteriormente. La curva de calibración se construyó
graficando la relación cociente de áreas (área
CP-60,300/área Til) frente a la concentración
de tulatromicina. El rango de concentración
(mg/kg) fue: 0,48 – 12,96 para hígado, 0,53
– 13,43 pasa riñón, 0,09 – 1,27 para músculo,
1,08 – 25,33 para sitio de inyección y 0,01 –
0,37 para grasa. Para el límite de cuantificación
(LOQ), se considera el primer punto de la curva de calibración que cumpla con la exactitud y
precisión establecidas. Criterio de aceptación:
cada punto de la curva debe cumplir con los valores de exactitud.
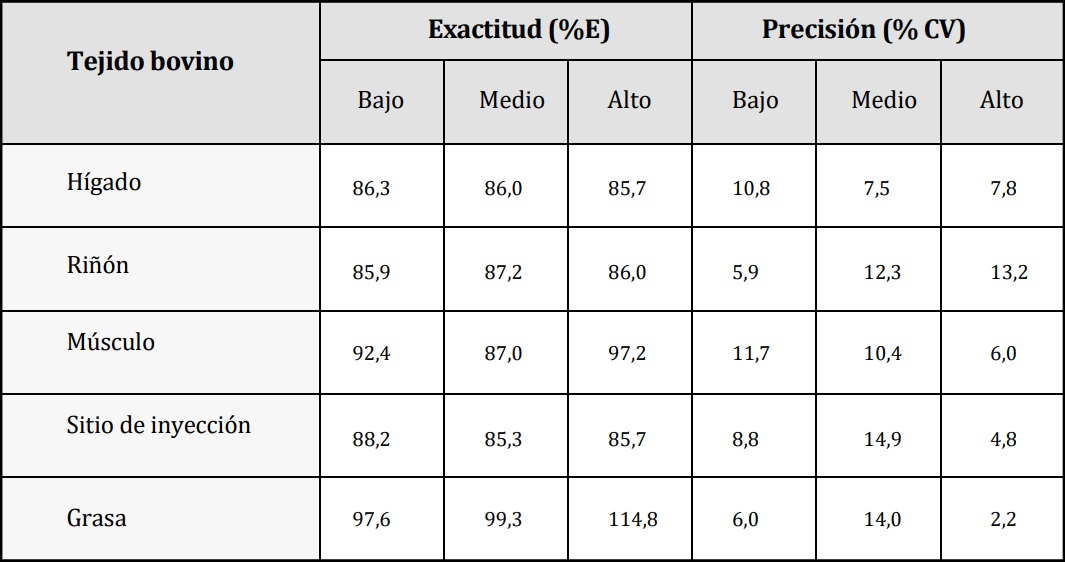
Exactitud y precisión. Se estudiaron por
análisis triplicado y quintuplicado, respectivamente, en los niveles bajo, medio y alto del rango
analítico de trabajo, siendo para hígado 0,97-
5,50-12,92, para riñón 1,00-5,76-13,39, para
músculo 0,15-0,60-1,27, para sitio de inyección
1,99-12,60-24,66 y para grasa 0,5-0,22-0,30 mg/
kg. La exactitud se calcula a partir de la concentración obtenida respecto de la concentración
real y se expresa como porcentaje (% E), y la
precisión como porcentaje de coeficiente de variación (% CV). Criterio de aceptación: la exactitud debe estar incluida en el rango 70-110%
o 80-120%. La precisión < 15 %, excepto para
el LOQ < 20%.
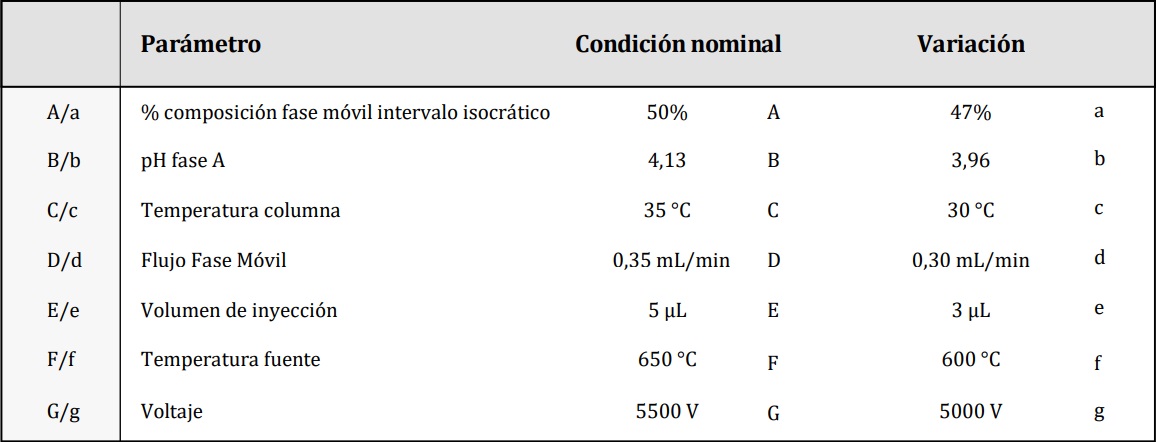
Robustez. Se realizaron pequeños cambios de parámetros que pueden tener influencia
significativa sobre los resultados. Se utilizó el diseño de Placket y Burman que permite estudiar
n variables en n+1 ensayos. Los parámetros estudiados fueron: % composición fase móvil, pH
fase A, temperatura columna, flujo fase móvil, volumen de inyección, temperatura fuente y voltaje
de ionización por electrospray (tabla 2).
Tabla 2. Parámetros estudiados para robustez.
Estabilidad. Se determinó la estabilidad química del analito a largo plazo (> seis meses) en cada una de las matrices de bovino. Se estudió por análisis triplicado de las muestras fortificadas en los niveles de concentración bajo, medio y alto del rango analítico de trabajo, (como se mencionó previamente en exactitud y precisión), almacenamiento a -80 °C, y se calculó la precisión y exactitud.
RESULTADOS
En primer lugar, se procedió a validar la metodología analítica en los diferentes tejidos bovinos. La técnica empleada fue selectiva, dado que se observó para cada una de las matrices ausencia de interferencia en el compuesto de interés y el EI propuesto. También resultó ser robusto porque los parámetros estudiados no tuvieron influencia significativa sobre el resultado final de los análisis. En la tabla 3 se muestran los resultados de las cifras de méritos estudiadas. Para cada tejido, en el nivel de estudio se informan: rango analítico, LOQ, % E y % CV; cumpliendo los criterios de aceptación establecidos por organismos de referencia en la temática. En la Figura 2, se grafican las curvas de calibrado para cada tejido bovino. En cuanto al estudio de estabilidad a largo plazo, el compuesto se mantuvo estable en las condiciones de almacenamiento propuesta para las diferentes matrices de bovino en estudio (tabla 4).
Tabla 3. Cifras de mérito estudiadas en tejidos bovinos.
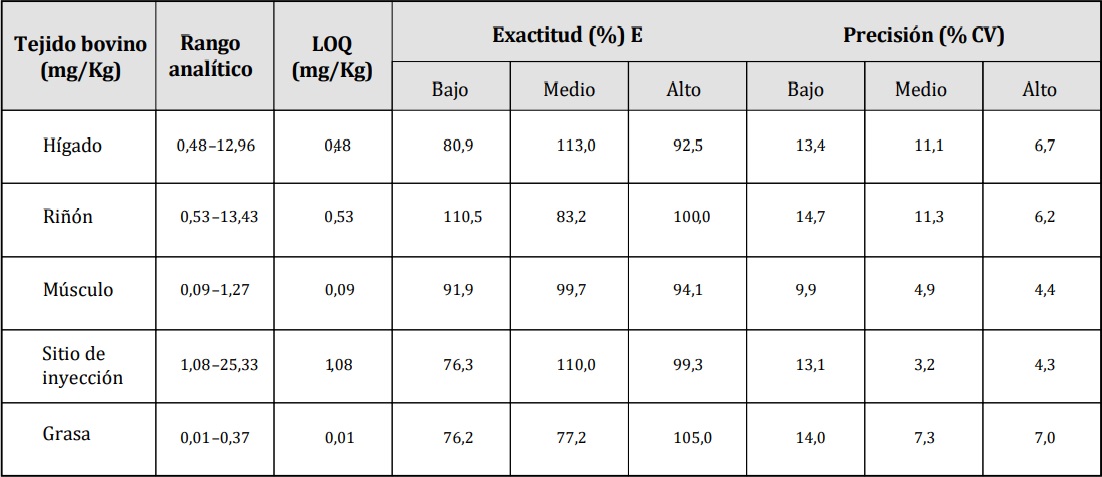
LOQ: límite de cuantificación; % CV: coeficiente de variación.
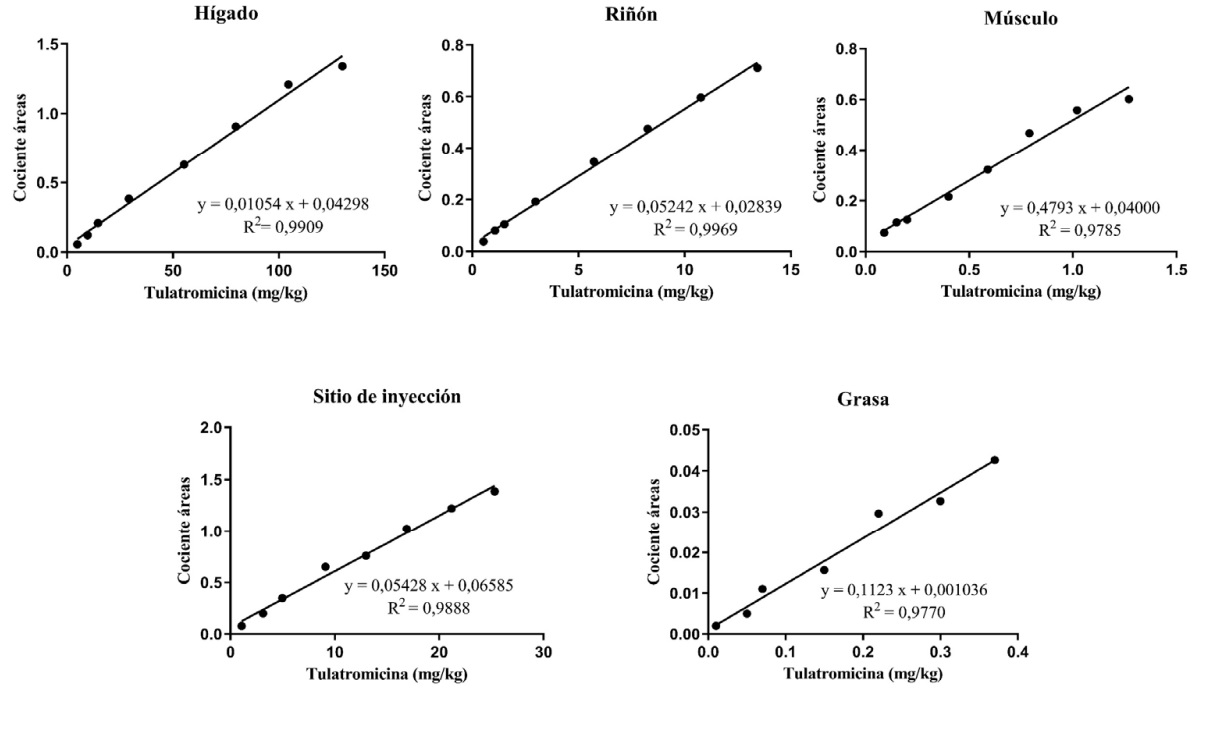
Figura 2. Curva de calibrado en los diferentes tejidos de bovinos
Tabla 4. Estabilidad a largo plazo (> 6 meses) en tejidos bovinos a -80 ºC en diferentes concentraciones.
% CV: coeficiente de variación.
DISCUSIÓN Y CONCLUSIONES
El método analítico propuesto mediante LC ESI MS/MS proporciona un procedimiento simple, rápido y confiable para la identificación y cuantificación de CP-60,300 en tejidos bovinos. La determinación analítica de CP-60,300 es el método regulatorio aprobado por los organismos reguladores y está diseñado para detectar concentraciones de cualquier isoforma de tulatromicina. En el tratamiento analítico propuesto, la etapa de hidrolisis de los tejidos en estudio asegura convertir la tulatromicina y sus metabolitos en el residuo marcador (CP-60,300), correlacionándose luego en la curva de calibración la respuesta con la cantidad presente de tulatromicina según la siguiente ecuación:
Por lo tanto, su validación constituye
una herramienta de gran utilidad para la cuantificación del analito de interés en tejidos de
bovinos, el conocimiento sobre su acumulación
y metabolización, y la posibilidad de determinar los tiempos de retiro de los animales de
producción para ajustarse a los LMR en los tejidos comestibles, garantizando la seguridad e
inocuidad de los alimentos sin inconvenientes
para la salud pública.
Múltiples estudios clínicos muestran
que la tulatromicina es eficaz en el tratamiento
de enfermedades respiratorias en bovinos. Por
otra parte, otros trabajos muestran un efecto inmunomodulador sobre macrófagos4. A su vez, al
igual que la tilmicosina, la tulatromicina se ha
desarrollado para la administración parenteral
única en bovinos, lo que minimiza la manipulación de los animales y maximiza el cumplimiento de los tratamientos. A raíz de estos resultados, la tulatromicina ha sido registrada en
varios países para tratar afecciones de importancia económica, particularmente problemas
respiratorios.1,14. A su vez, una característica
farmacocinética importante de la droga es su
amplia distribución en los tejidos y la buena penetración celular, logrando mayores concentraciones en tejidos que en plasma de bovinos. De
esta forma, asegurar la ausencia de residuos en
los mismos o tratar que el residuo no supere los
LMR resulta fundamental1. Según la Agencia Europea de Medicamentos (EMA)6 detalla que la
ingesta diaria admisible (IDA) de tulatromicina
en humanos es de 3 mg/kg de peso. En este sentido, se pudo observar que en el día 15 post-administración de tulatromicina la concentración
del analito se encontró por debajo del IDA. Por
lo tanto, esta metodología analítica, que cumple
con los criterios de aceptación establecidos por
los organismos de cada país, resulta adecuada
para el análisis de tulatromicina y la determinación de los tiempos de retiro de los animales.
1. Benchaoui HA., Nowakowski M., Sherington J., Rowan
TG., Sunderland SJ. Pharmacokinetics and lung tissue
concentrations of tulathromycin in swine. J. Vet. Pharmacol. Therap. 2004; 27(4):203–10.
2. Boner PL., Gottschall DW., Kim-Kang H. Determination
and confirmation of tulathromycin residues in bovine
liver and porcine kidney via their common hydrolytic
fragment using high-performance liquid chromatography/tandem mass spectrometry. J AOAC Int. 2011;
94(2):436–45.
3. Codex Alimentarius Volume 3 “Residues of Veterinary
Drugs in Foods”, 2nd ed., Joint FAP/WHO Food Standards Program, FAO, Rome Italy. 1993.
4. Desmonts de Lamache D., Mogues R., Siddiq A., et al.
Immuno-modulating properties of Tulathromycin in
porcine monocyte-derived macrophages infected with
porcine reproductive and respiratory syndrome virus,
PLoS One 2019; 14 (8).
5. European Commission, Commission Regulation (EU)
No 37/2010 of 22 December 2009 on pharmacologically active substances and their classification regarding
maximum residue limits in foodstuffs of animal origin,
Off. J. Eur. Union. 2010;1–72.
6. European Medicines Agency: Committee for Medicinal
Products for Veterinary Use. European public MRL assessment report (EPMAR) for tulathromycin – modification of the ADI and MRLs in bovine and porcine species,
EMA/CVMP/380257/2014.
7. FDA, Guidelines for the Validation of Chemical Methods
in Food, Feed, Cosmetics, and. Veterinary Products, 3rd
Edition. 2019.
8. Instrucción Normativa No. 51 de 19 de diciembre de
2019. Agencia Nacional de Vigilancia Sanitaria (ANVISA). Diário Oficial da União. 2019. 249:98
9. List of Maximum Residue Limits (MRLs) for Veterinary
Drugs in Foods. Agencia del gobierno de Canadá. 2021.
10. Ministério da Agricultura, P. e A. (MAPA). Requisitos e
critérios específicos para funcionamento dos laboratórios de análises de resíduos e contaminantes em alimentos integrantes da rede nacional de laboratórios agropecuários. Angew. Chemie Int. Ed. 6(11). 2009.
11. Resolución-559-2011 – Servicio Nacional de Sanidad y
Calidad Agroalimentaria (SENASA).
12. Saito-Shida S., Kashiwabara N., Nemoto S., Akiyama H.
Determination of the total tulathromycin residues in
bovine muscle, fat, and liver by liquid chromatography-tandem mass spectrometry, J Chromatogr B Analyt
Technol Biomed Life Sci. 2019; 1110–1111:51–58.
13. Sanco/12571/2013. Guidance document on analytical
quality control and validation procedures for pesticide
residues analysis in food and feed. European Commission Health & Consumer Protection Directorate-General.
14. Villarino N., Brown SA., Martin-Jiménez T. The role of the
macrolide tulathromycin in veterinary medicine, Vet. J.
2013; 198 (2):352–7.
15. Wang X., Tao YF., Huang LL., et al. Pharmacokinetics of
tulathromycin and its metabolite in swine administered
with an intravenous bolus injection and a single gavage,
J. Vet. Pharmacol Ther. 2012; 35(3):282–9.
